Event submissions
EventsMOL2NET'17, Conference on Molecular, Biomed., Comput. & Network Science and Engineering, 3rd ed.
Published

This submission belongs to the session 06. NICEXSM-03: North-Ibero-American Congress on Exp. & Simul. Methods, Valencia, Bilbao, Spain-Paraiba, Brasil-Miami, USA, 2017 of the event MOL2NET'17, Conference on Molecular, Biomed., Comput. & Network Science and Engineering, 3rd ed.
Published date
04 Dec, 2017
Citation
Luciana Scotti, Jéssika Viana, Marcus T Scotti, Molecular docking studies of benzothiazinone derivatives in the search for new tuberculostatic agents, in Proceedings of MOL2NET'17, Conference on Molecular, Biomed., Comput. & Network Science and Engineering, 3rd ed., 15 January–15 December 2017, MDPI: Basel, Switzerland, doi: 10.3390/mol2net-03-05052
Share
Email
Facebook
Twitter
LinkedIn
Molecular docking studies of benzothiazinone derivatives in the search for new tuberculostatic agents

Jéssika Viana 1

Marcus T Scotti 1

Luciana Scotti 2
1. Federal University of Paraiba
2. Federal University of Paraiba, Brazil
Abstract
Despite the efforts to reduce the rate of infection, the resurgence of Tuberculosis, accompanied by strains of Mycobacterium tuberculosis, calls attention to the search for new drugs that can bypass resistance mechanisms. Among the strategies for the development of antitubercular lead compounds, benzothiazinones (BTZ) are included, with highly selective mechanism of action of DprE1 flavoenzyme. We studied four compounds in the antituberculosis activity (IC50 values between 0.029 - 0.060 μg/ml). These derivatives were subjected to energy minimization and Molecular Docking calculations in the software Molegro Virtual Docker, from which the binding free energy calculations showed that the suggested compounds had better binding affinity with DprE1 when compared to PBTZ169, a crystallized inhibitor of DprE1. Our results showed that the compounds showed similar crucial binding interactions between the compounds, which may determine that there was molecular rearrangement within the active site itself. Thus, our studies evidenced the probable interactions that favor the activity of the derivatives, as well as other interactions of the compounds with other residues not reported in the literature, and these may act as lead compounds in the development of new antituberculosis drugs.
Keywords
Tuberculosis
benzothiazinones
Docking
Poster
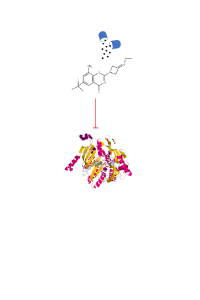
pres jes.pdf
Applications of Structure Based Drug Design Approaches towards Design and Development of Calcium Channel Blockers
The KP algorithm for the analysis of the optimal flow of information